02/07/2018
La vida, tal como la conocemos, depende de la existencia de compartimentos separados, y en el centro de esta organización se encuentran las membranas biológicas. Estas estructuras delicadas y dinámicas actúan como barreras selectivas, controlando el paso de sustancias y manteniendo el equilibrio interno de las células y los órganos. Sin embargo, ¿qué ocurre cuando esta barrera pierde su selectividad y su permeabilidad aumenta de forma descontrolada? Las consecuencias pueden ser profundas y, a menudo, devastadoras para la función celular y la salud en general. Este artículo explorará los intrincados efectos de un aumento en la permeabilidad de dos tipos de membranas cruciales: la membrana mitocondrial y la membrana alveolo-capilar, revelando cómo un desequilibrio en su función puede desencadenar una cascada de eventos perjudiciales.
Desde la regulación energética de nuestras células hasta el vital intercambio de gases en nuestros pulmones, la integridad de las membranas es fundamental. Entender los mecanismos y las repercusiones de una permeabilidad alterada nos ofrece una visión fascinante de la complejidad de la fisiología humana y de las enfermedades que surgen cuando estos procesos se ven comprometidos.
- La Permeabilidad Mitocondrial: Un Equilibrio Vital para la Supervivencia Celular
- La Permeabilidad de la Membrana Alveolo-Capilar: Un Intercambio Crucial para la Vida
- Tabla Comparativa: Consecuencias del Aumento de Permeabilidad
- Preguntas Frecuentes sobre la Permeabilidad de las Membranas
- ¿Por qué es importante la permeabilidad de las membranas celulares?
- ¿Puede la permeabilidad mitocondrial mejorarse o controlarse?
- ¿Cómo se diagnostica el aumento de la permeabilidad alveolo-capilar?
- ¿Qué relación tiene el estrés oxidativo con la permeabilidad de las membranas?
- ¿Es siempre dañino un aumento de la permeabilidad?
- Conclusión
La Permeabilidad Mitocondrial: Un Equilibrio Vital para la Supervivencia Celular
Las mitocondrias, a menudo llamadas las 'centrales energéticas' de la célula, son orgánulos esenciales que producen la mayor parte del ATP (adenosín trifosfato), la moneda energética celular, a través de un proceso llamado fosforilación oxidativa. Para que este proceso funcione correctamente, la membrana interna mitocondrial debe mantener una permeabilidad altamente regulada, creando un gradiente electroquímico necesario para la síntesis de ATP. Sin embargo, en ciertas condiciones de estrés, como la isquemia (falta de flujo sanguíneo) seguida de reperfusión (restablecimiento del flujo), esta permeabilidad puede aumentar drásticamente, lo que lleva a un daño celular significativo.
El Poro de Transición de la Permeabilidad Mitocondrial (PTP)
Uno de los mecanismos más críticos que alteran la permeabilidad de la membrana mitocondrial es la formación del Poro de Transición de la Permeabilidad Mitocondrial (PTP). Este poro es un canal de alta conductancia dependiente de voltaje, ubicado en la membrana mitocondrial interna. Su apertura es un evento complejo y multifacético, mediado por diversos factores que, en última instancia, conducen a una alteración masiva de la permeabilidad.
Mecanismos de Apertura y Consecuencias Celulares
La apertura del PTP puede ser desencadenada por una serie de mecanismos. Se ha observado que alteraciones en el microambiente lipídico de la membrana y la inactivación de la proteína BCL2 preceden a la formación del PTP. Es importante destacar que la formación del PTP requiere un pH elevado, lo que explica por qué ocurre predominantemente durante la fase de reperfusión después de un evento isquémico, cuando el pH intracelular tiende a aumentar.
Cuando el PTP se forma y se abre, permite una entrada masiva de agua al interior de la mitocondria, lo que provoca su hinchazón osmótica. Esta hinchazón desestabiliza la membrana mitocondrial, conduciendo a su disfunción. La modificación de proteínas estructurales de la membrana mitocondrial, como los tioles de ANT (translocasa de nucleótidos de adenina), la ciclofilina D (Cyp D) y VDAC1 (canal aniónico dependiente de voltaje), también contribuye a la apertura del PTP. La apertura de este poro interrumpe directamente la generación de ATP, ya que VDAC facilita el transporte de ATP/ADP a través de la membrana mitocondrial externa, y el grupo ANT lo hace a través de la membrana mitocondrial interna. Por su parte, CypD, una proteína de la matriz mitocondrial, asiste en los cambios conformacionales de otras proteínas. La activación de Cyp D por factores inductores de apoptosis (AIF y AMID) también inicia la formación del PTP.
Las proteínas de la familia BCL2 (como Bax, Bid, Puma y BNIP3) también desempeñan un papel crucial al aumentar la permeabilización de las membranas mitocondriales interna y externa, lo que conduce a la formación del poro de permeabilidad. La reperfusión tras la isquemia intensifica la activación de las proteínas de la familia BCL2 y regula al alza otros factores apoptóticos. Además, la presencia de agentes oxidantes, fosfato inorgánico (Pi) y un pH alcalino favorecen la formación del PTP. La disrupción de la barrera de permeabilidad de la membrana mitocondrial interna por el PTP desencadena el desacoplamiento de la fosforilación oxidativa, la hinchazón osmótica y la ruptura de la membrana externa. La liberación concomitante de proteínas mitocondriales, como el citocromo c, activa la cascada de caspasas, lo que a su vez induce endonucleasas, llevando finalmente a un daño irreversible del ADN y a la muerte celular, particularmente la muerte neuronal en el contexto isquémico.
Es relevante mencionar que el modo de formación del PTP en la población mitocondrial determina si la muerte celular será necrótica o apoptótica. Por ejemplo, la depleción de la creatina quinasa mitocondrial-1 (CKMT1) lleva a la muerte neuronal apoptótica, y la proteína p53 por sí sola induce la apoptosis, mientras que la interacción de p53 con CypD conduce a la muerte neuronal necrótica. La hinchazón de la matriz mitocondrial que precede al desacoplamiento de la fosforilación oxidativa provoca la liberación del citocromo c, el cual activa AIF y la endonucleasa G (Endo G). Estas se translocan al núcleo, activando la condensación de la cromatina y la fragmentación del ADN. El citocromo c también activa los receptores IP3, aumentando el calcio citosólico y la generación de ROS (especies reactivas de oxígeno). Además, el PTP induce la fisión/fusión mitocondrial y la mitofagia, un proceso de autofagia que elimina mitocondrias dañadas. Así, la permeabilidad de la membrana mitocondrial es un fenómeno estrictamente regulado, con consecuencias importantes que pueden llevar a la muerte celular, especialmente la muerte neuronal en el contexto de una lesión isquémica.
El Estrés Oxidativo y Nitrooxidativo: Daño Generalizado
Más allá de la formación directa de poros, el aumento de la permeabilidad mitocondrial está intrínsecamente ligado al estrés oxidativo y nitrooxidativo, fenómenos que exacerban el daño celular y contribuyen a la muerte neuronal tras una lesión isquémica y de reperfusión.
El Impacto del Estrés Oxidativo
El deterioro mitocondrial debido a la isquemia y reperfusión genera un exceso de especies reactivas de oxígeno (ROS), lo que conduce a estrés oxidativo. Esto es una consecuencia de la excitotoxicidad por glutamato y se agrava por el aumento de la liberación de calcio mitocondrial, un factor clave en la progresión del daño isquémico cerebral. El aumento de calcio intracelular desencadena la oxidación de NADPH, que es crucial para la resistencia mitocondrial al eliminar los radicales libres. La oxidación de NADPH agrava el estrés oxidativo al reducir la actividad de eliminación de superóxidos y aumentar su acumulación. La reducción de la glutatión transferasa (GSH) también contribuye a la oxidación de NADPH, lo que a su vez acelera la muerte mitocondrial.
El daño oxidativo del fósforo inorgánico mitocondrial (Pi) también favorece la formación del PTP al generar enoles que producen aldehídos tripletes, que son a su vez especies reactivas de oxígeno. Además, la regulación al alza de las moléculas de adhesión celular (CAMs) provoca la infiltración de leucocitos y la liberación de ROS, exacerbando el daño isquémico. Enzimas como la xantina oxidasa, la ciclooxigenasa, la NADPH oxidasa leucocitaria, la superóxido dismutasa (SOD) y la eNOS desacoplada actúan como fuentes putativas de generación de ROS durante el estrés oxidativo, lo que lleva a la lesión neuronal. El daño directo de proteínas, lípidos, ácidos nucleicos y otros componentes macromoleculares de la célula es la primera consecuencia. Las ROS también inducen la muerte neuronal dependiente e independiente de caspasas, y conducen a la pérdida del potencial mitocondrial, degenerando el axón a través de Sarm1.
Las ROS (radicales hidroxilo) también reaccionan con la β-Amiloide (Aβ), un activador de quinasa que protege las células del estrés oxidativo, lo que contribuye a la muerte neuronal. Indirectamente, las ROS desencadenan la liberación de citocromo c de la membrana mitocondrial mediante la translocación de p53, que controla la expresión de genes pro-apoptóticos como Bax, Bid y PUMA. El estrés oxidativo también media la apoptosis y la muerte celular inducida por ROS a través de vías independientes de caspasas, como la vía dependiente de calpaína. Otra vía de daño oxidativo es la activación del factor de transcripción FOXO3. La inhibición de FOXO3 por el factor de crecimiento similar a la insulina-I (IGF-I) durante la isquemia también provoca la generación de ROS.
La barrera hematoencefálica (BHE) es una puerta crucial que se ve afectada por los inductores del estrés oxidativo en las primeras etapas de diferentes enfermedades cerebrales. Las ROS activan las pro-metaloproteinasas de matriz (proMMP), lo que conduce a la degradación de la matriz neurovascular y, en última instancia, a la destrucción de la BHE. Además, bajo condiciones de estrés oxidativo, el factor de transcripción NF-κB se une a la región promotora de MMP-9, lo que resulta en la degradación de colágenos y lamininas en la lámina basal, alterando la integridad de la pared vascular y la permeabilidad de la BHE.
El estrés oxidativo también inhibe la actividad del proteasoma, el complejo proteico responsable de la degradación de proteínas. Esta inhibición inicia aún más la activación de caspasas, lo que lleva a la muerte neuronal apoptótica. También se observan niveles elevados de proteínas ubiquitinadas durante la isquemia, especialmente 4-hidroxinonenal (4-HNE), un subproducto de la peroxidación lipídica que inhibe la actividad del proteasoma, altera la bicapa fosfolipídica y reacciona con enzimas mitocondriales, lo que lleva a un fallo energético mitocondrial. La peroxidación lipídica también puede dañar la membrana de los glóbulos rojos. Las lipoproteínas de baja densidad oxidadas (ox-LDL) actúan como señales de estrés y juegan un papel influyente en la permeabilidad de la BHE, activando MMP2 y MMP9 y desestabilizando la BHE.
El Peligro del Estrés Nitrooxidativo
El estrés nitrooxidativo es otro factor crucial en el daño celular, especialmente en el sistema nervioso. Las enzimas óxido nítrico sintasa neuronal (nNOS) e inducible (iNOS), expresadas en neuronas, células gliales y miocitos vasculares durante la isquemia/reperfusión, contribuyen a la liberación de óxido nítrico (NO) de las células endoteliales. iNOS, activada por interleucinas y TNF-α, produce una cantidad de NO de larga duración. Cuando el NO reacciona con el radical superóxido, forma peroxinitrito (ONOO-), una especie reactiva de nitrógeno altamente dañina. El aumento de peroxinitrito después de la isquemia afecta drásticamente la supervivencia de las células cerebrales, lo que lleva a la muerte neuronal.
La acumulación de especies reactivas de nitrógeno conduce al agotamiento de la actividad de glutatión y tiorredoxina, lo que resulta en disfunción mitocondrial. Además, los macrófagos infiltrados y la microgliosis agravan la generación de especies reactivas de nitrógeno. El peroxinitrito y otras especies reactivas de nitrógeno son perjudiciales para proteínas, lípidos y tanto el ADN neuronal como el mitocondrial. Provocan peroxidación lipídica, oxidación de proteínas y daño al ADN. También inducen varios factores de transcripción que conducen a la inflamación mediada por citoquinas y perturbaciones epigenéticas que exacerban la respuesta inflamatoria mediada por el factor nuclear kappa-B (NF-κB). El peroxinitrito provoca la nitrotirosinación de proteínas, formando 3-nitrotirosina, una modificación postraduccional patológica que altera el funcionamiento fisiológico de las proteínas. Aunque el peroxinitrito tiene una vida corta, su capacidad para difundirse a través de las membranas biológicas extiende su efecto dañino a las células y tejidos vecinos.
La Apoptosis y la Comunicación ER-Mitocondrial
El aumento de la permeabilidad mitocondrial y el estrés asociado están íntimamente ligados a la apoptosis, el proceso de muerte celular programada, y a la intrincada relación entre las mitocondrias y el retículo endoplasmático (RE).
La Vía de la Apoptosis
El citocromo c (cyt c) liberado de las mitocondrias activa AIF, procaspasa-9 y caspasa-3, desencadenando la apoptosis mediante la formación del apoptosoma. Aunque los mecanismos exactos de la liberación de citocromo c no se comprenden del todo, se han revelado dos posibles vías: el daño mecánico de la membrana mitocondrial externa debido a la hinchazón de la matriz, y la translocación de la proteína pro-apoptótica Bax a la membrana mitocondrial. La interacción compleja de proteínas anti y pro-apoptóticas con la transición de permeabilidad mitocondrial (MPT) sugiere que la MPT es una parte integral de la apoptosis. El citocromo c oxidado activa rápidamente las caspasas-9. La citocromo oxidasa mitocondrial oxida el citocromo c externo, y varios mecanismos aceleran la oxidación del citocromo c, lo que lleva a la apoptosis. La reducción citosólica de citocromo c por las neuronas es alta y depende del estado redox del glutatión intracelular.
El citocromo c también induce el gen tumoral p53, un modulador clave de las respuestas al estrés celular. El gen p53 desencadena la apoptosis mediante la activación de genes pro-apoptóticos y mecanismos independientes de la transcripción en respuesta a estímulos de muerte. El p53 puede alterar la permeabilidad de la membrana mitocondrial externa formando un complejo inhibidor con la proteína de la familia Bcl-2, lo que lleva a la liberación de citocromo c.
La Interacción entre Retículo Endoplasmático y Mitocondrias
Desarrollos recientes han revelado que el estrés del retículo endoplasmático (RE) es un evento de señalización esencial para la lesión neuronal resultante de la isquemia/reperfusión (I/R). Ciertos estímulos como la isquemia, la hipoxia y la hipertensión pueden desencadenar la acumulación de proteínas desplegadas en el lumen del RE, lo que lleva a la respuesta a proteínas desplegadas (UPR). Esta respuesta implica la expansión de las membranas del RE, la degradación acelerada de proteínas desplegadas, el aumento de la traducción de chaperonas de plegamiento y la inhibición de otras síntesis de proteínas. Tras el estrés del RE inducido por la privación de oxígeno y glucosa (OGD), el RE se hincha. Un mayor volumen del RE permite que un mayor número de proteínas mal plegadas se incorporen a la membrana del RE, reduciendo la concentración de intermediarios proteicos y aumentando la capacidad celular para lidiar con el abundante daño proteico.
La isquemia cerebral puede perturbar la función del RE, lo que resulta en la acumulación de proteínas desplegadas en el lumen del RE, una condición conocida como estrés del RE. El estrés oxidativo mitocondrial que se desarrolla durante la isquemia contribuye sustancialmente a la muerte neuronal. Al igual que las mitocondrias, el retículo endoplasmático (RE) es otro orgánulo celular que funciona en asociación con las mitocondrias. El calcio intracelular del RE juega un papel significativo en el mantenimiento de la homeostasis del RE y la comunicación con las mitocondrias (interacción RE-mitocondrias). El aumento del calcio intracelular conduce al estrés del RE y a la apertura del poro de transición mitocondrial, lo que a su vez provoca la liberación de citocromo c, lo que gradualmente lleva a la activación de factores apoptóticos. Las membranas asociadas a las mitocondrias también ayudan en la comunicación entre el RE y las mitocondrias. El daño mitocondrial debido al aumento del calcio intracelular conduce a la fragmentación mitocondrial y al desacoplamiento del RE, lo que resulta en la inhibición de la liberación de calcio del RE, lo que a su vez conduce a la apoptosis. El estrés oxidativo y del RE después de la isquemia también inducen la mitofagia (autofagia), que inicialmente previene la necrosis y la apoptosis al mantener la homeostasis iónica. Sin embargo, el aumento del estrés autofágico conduce a una activación lisosomal masiva y, en última instancia, a la muerte celular. Así, la mitocondria parece desempeñar un papel significativo en la orquestación de la muerte neuronal tras la isquemia y la reperfusión.
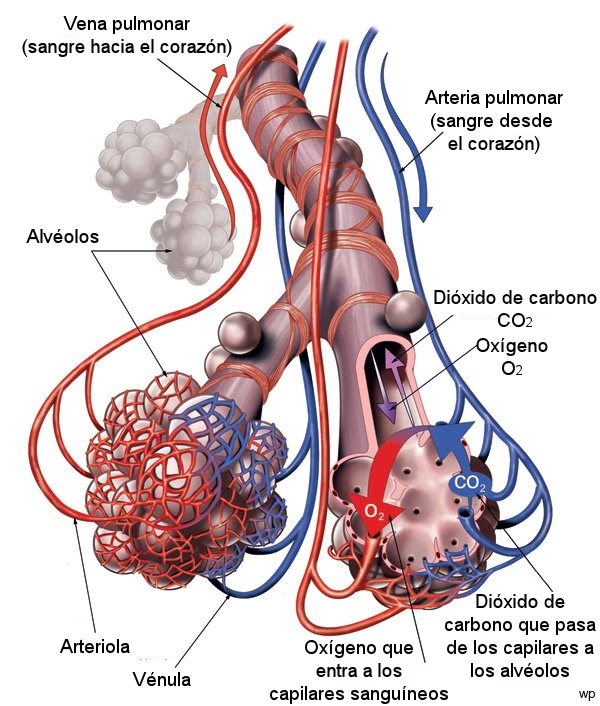
La Permeabilidad de la Membrana Alveolo-Capilar: Un Intercambio Crucial para la Vida
Mientras que la permeabilidad mitocondrial se relaciona con la energía y la muerte celular a nivel subcelular, la permeabilidad de la membrana alveolar-capilar es fundamental para la función pulmonar y el suministro de oxígeno a todo el cuerpo. Esta membrana es la interfaz donde se produce el intercambio de gases vital para la respiración.
Función Normal de la Membrana Alveolo-Capilar
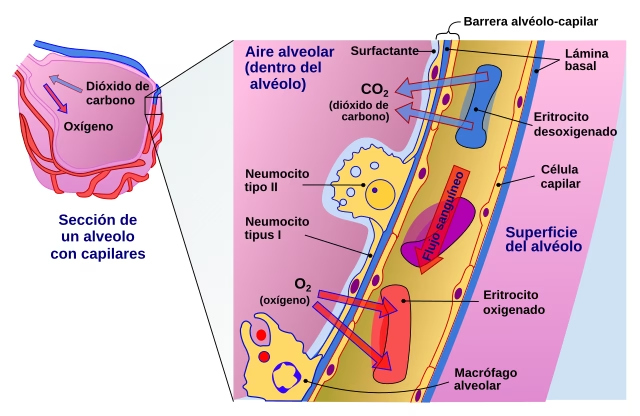
La función principal del aparato respiratorio es inhalar oxígeno y eliminar dióxido de carbono. El oxígeno inhalado penetra en los pulmones y alcanza los alvéolos, que son pequeños sacos de aire. Las capas de células que revisten los alvéolos y los capilares circundantes se disponen ocupando el espesor de una sola célula y están en contacto estrecho unas con otras. Esta barrera entre el aire y la sangre tiene un grosor aproximado de 1 micra (1/1000 de milímetro o 0,00004 pulgadas). El oxígeno atraviesa rápidamente esta barrera aire-sangre y llega hasta la sangre que circula por los capilares. Igualmente, el dióxido de carbono pasa de la sangre al interior de los alvéolos, desde donde es exhalado al exterior.
La sangre oxigenada circula desde los pulmones por las venas pulmonares y, al llegar al lado izquierdo del corazón, es bombeada hacia el resto del organismo. La sangre con déficit de oxígeno y cargada de dióxido de carbono vuelve al lado derecho del corazón a través de las venas cavas. A continuación, la sangre es impulsada a través de la arteria pulmonar hacia los pulmones, donde recoge el oxígeno y libera el dióxido de carbono.
Para mantener la absorción de oxígeno y la emisión de dióxido de carbono, entran y salen de los pulmones entre 5 y 8 litros de aire por minuto en reposo, y cada minuto se transfieren alrededor de 3/10 de litro de oxígeno desde los alvéolos hasta la sangre. Al mismo tiempo, un volumen similar de dióxido de carbono pasa de la sangre a los alvéolos y es exhalado. Durante el ejercicio, es posible respirar más de 100 L de aire por minuto y extraer de este aire 3 L de oxígeno por minuto. La velocidad de entrada del oxígeno en el organismo es una medida importante de la cantidad total de energía consumida por este.
Los tres procesos esenciales para la transferencia del oxígeno desde el aire del exterior a la sangre que fluye por los pulmones son: ventilación, difusión y perfusión. La ventilación es el proceso por el cual el aire entra y sale de los pulmones. La difusión es el movimiento espontáneo de gases entre los alvéolos y la sangre de los capilares pulmonares sin intervención de energía alguna o esfuerzo del organismo. La perfusión es el proceso por el cual el sistema cardiovascular bombea la sangre a los pulmones y otros órganos y tejidos del cuerpo. La circulación corporal es un vínculo esencial entre la atmósfera, que contiene oxígeno, y las células del cuerpo, que lo consumen.
¿Qué Sucede con el Aumento de la Permeabilidad Alveolar?
A diferencia de la permeabilidad mitocondrial que se altera en contextos de estrés celular, el aumento de la permeabilidad alvéolo-capilar en los pulmones es siempre una señal de daño. Este fenómeno indica una lesión en el epitelio respiratorio, la delicada capa de células que recubre los alvéolos. Cuando este epitelio se daña, su capacidad de barrera se ve comprometida, lo que permite un aumento anómalo en el paso de fluidos, proteínas e incluso células desde los capilares sanguíneos hacia el espacio alveolar. Esto puede conducir a la acumulación de líquido en los alvéolos (edema pulmonar), dificultando gravemente el intercambio de gases. En lugar de un eficiente paso de oxígeno y dióxido de carbono, el espacio alveolar se llena de elementos que obstruyen la difusión, lo que lleva a hipoxemia (niveles bajos de oxígeno en la sangre) y a una insuficiencia respiratoria. Esta condición es característica de diversas patologías pulmonares agudas, como el síndrome de dificultad respiratoria aguda (SDRA), donde la inflamación y el daño tisular comprometen severamente la función pulmonar.
Tabla Comparativa: Consecuencias del Aumento de Permeabilidad
| Característica | Permeabilidad Mitocondrial Aumentada | Permeabilidad Alveolo-Capilar Aumentada |
|---|---|---|
| Contexto principal | Estrés celular (isquemia/reperfusión) | Daño pulmonar agudo (inflamación, lesión) |
| Ubicación | Membranas interna y externa de las mitocondrias | Membrana entre alvéolos y capilares pulmonares |
| Mecanismo clave | Formación del Poro de Transición de la Permeabilidad (PTP) | Daño al epitelio respiratorio |
| Sustancias que atraviesan | Agua, iones, proteínas mitocondriales (citocromo c, AIF) | Fluidos, proteínas, células sanguíneas |
| Consecuencias directas | Hinchazón mitocondrial, desacoplamiento de fosforilación oxidativa, liberación de citocromo c, ATP depleción | Edema pulmonar (líquido en alvéolos), deterioro del intercambio de gases |
| Impacto celular/orgánico | Muerte celular (apoptosis, necrosis), disfunción neuronal, daño cerebral | Insuficiencia respiratoria, hipoxemia sistémica |
| Factores contribuyentes | Estrés oxidativo, estrés nitrooxidativo, calcio intracelular elevado, disfunción de proteínas (CypD, VDAC, ANT), BCL2 | Inflamación, infecciones, toxinas, trauma |
Preguntas Frecuentes sobre la Permeabilidad de las Membranas
¿Por qué es importante la permeabilidad de las membranas celulares?
La permeabilidad de las membranas celulares es vital porque permite a la célula mantener un ambiente interno estable (homeostasis), a pesar de las fluctuaciones en su entorno. Regula el paso selectivo de nutrientes, iones y moléculas de señalización, al tiempo que previene la entrada de sustancias dañinas y la fuga de componentes celulares esenciales. Este control es fundamental para todos los procesos biológicos, desde la producción de energía hasta la comunicación celular y la respuesta a estímulos.
¿Puede la permeabilidad mitocondrial mejorarse o controlarse?
Sí, existen vías de investigación y posibles estrategias para modular la permeabilidad mitocondrial, especialmente en el contexto de enfermedades. Por ejemplo, se estudian fármacos que pueden inhibir la apertura del Poro de Transición de la Permeabilidad (PTP), como la ciclosporina A, o compuestos que reducen el estrés oxidativo. La comprensión de los mecanismos moleculares que regulan el PTP ofrece la esperanza de desarrollar terapias que puedan proteger las células del daño en condiciones como la isquemia y la reperfusión.
¿Cómo se diagnostica el aumento de la permeabilidad alveolo-capilar?
El aumento de la permeabilidad alveolo-capilar se diagnostica mediante una combinación de hallazgos clínicos, radiológicos y, a veces, pruebas de laboratorio. Los síntomas incluyen dificultad para respirar, tos y niveles bajos de oxígeno. Las radiografías o tomografías computarizadas de tórax pueden mostrar edema pulmonar (líquido en los pulmones). En algunos casos, se pueden realizar pruebas más específicas, como el análisis del líquido de lavado broncoalveolar, para detectar la presencia de proteínas o células sanguíneas que no deberían estar en el espacio alveolar, confirmando el daño de la barrera.
¿Qué relación tiene el estrés oxidativo con la permeabilidad de las membranas?
El estrés oxidativo, causado por un desequilibrio entre la producción de especies reactivas de oxígeno (ROS) y la capacidad del cuerpo para neutralizarlas, puede dañar directamente los componentes de las membranas, como los lípidos y las proteínas. Este daño altera la estructura y la función de la membrana, aumentando su permeabilidad. En el caso de las mitocondrias, el estrés oxidativo es un potente inductor de la formación del Poro de Transición de la Permeabilidad (PTP), lo que lleva a la disfunción mitocondrial y la muerte celular. De manera similar, en los pulmones, las ROS pueden dañar el epitelio alveolo-capilar, aumentando su permeabilidad y causando edema pulmonar.
¿Es siempre dañino un aumento de la permeabilidad?
En el contexto biológico de las membranas celulares y de los órganos, un aumento descontrolado de la permeabilidad es casi siempre perjudicial. Indica una pérdida de la integridad de la barrera y de la selectividad, lo que lleva a la disfunción celular, la muerte celular o el daño orgánico, como se observa en la disfunción mitocondrial o el edema pulmonar. Sin embargo, en ciertos procesos fisiológicos, existen cambios temporales y controlados en la permeabilidad que son necesarios, como la permeabilidad transitoria en las uniones estrechas durante la inflamación para permitir el paso de células inmunes, pero estos son procesos regulados y no un aumento indiscriminado.
Conclusión
La permeabilidad de las membranas biológicas es un pilar fundamental de la vida. Su estricta regulación asegura el correcto funcionamiento de las células y los órganos, permitiendo los intercambios necesarios mientras mantiene la integridad interna. Sin embargo, cuando esta delicada regulación se pierde y la permeabilidad aumenta de forma descontrolada, las consecuencias son severas. Hemos explorado cómo el aumento de la permeabilidad mitocondrial, a través del PTP y en el contexto del estrés oxidativo y nitrooxidativo, conduce a la disfunción energética y la muerte celular, particularmente en el cerebro durante eventos isquémicos. Simultáneamente, el daño en la barrera alveolar-capilar pulmonar ilustra cómo un aumento de permeabilidad a nivel de órgano puede comprometer gravemente el intercambio vital de gases, llevando a insuficiencia respiratoria.
En ambos escenarios, la alteración de la permeabilidad actúa como un punto de inflexión, transformando orgánulos o tejidos funcionales en inductores de daño y enfermedad. Comprender estos mecanismos no solo profundiza nuestro conocimiento sobre la fisiología y patología, sino que también abre puertas para el desarrollo de futuras intervenciones terapéuticas destinadas a restaurar la homeostasis y proteger la salud celular y orgánica.
Si quieres conocer otros artículos parecidos a ¿Qué sucede cuando la permeabilidad de las membranas aumenta? puedes visitar la categoría Cabello.