23/08/2024
Los síndromes genéticos son afecciones que resultan de cambios en los genes de una persona, impactando su desarrollo y salud de diversas maneras. Aunque cada síndrome es único, comparten la particularidad de originarse en el material genético. En este artículo, exploraremos dos condiciones genéticas notables: el síndrome de Cornelia de Lange (CdLS) y el síndrome de Noonan, desglosando sus características distintivas, sus causas y cómo se manejan, con el objetivo de ofrecer una comprensión profunda y clara para quienes buscan información.
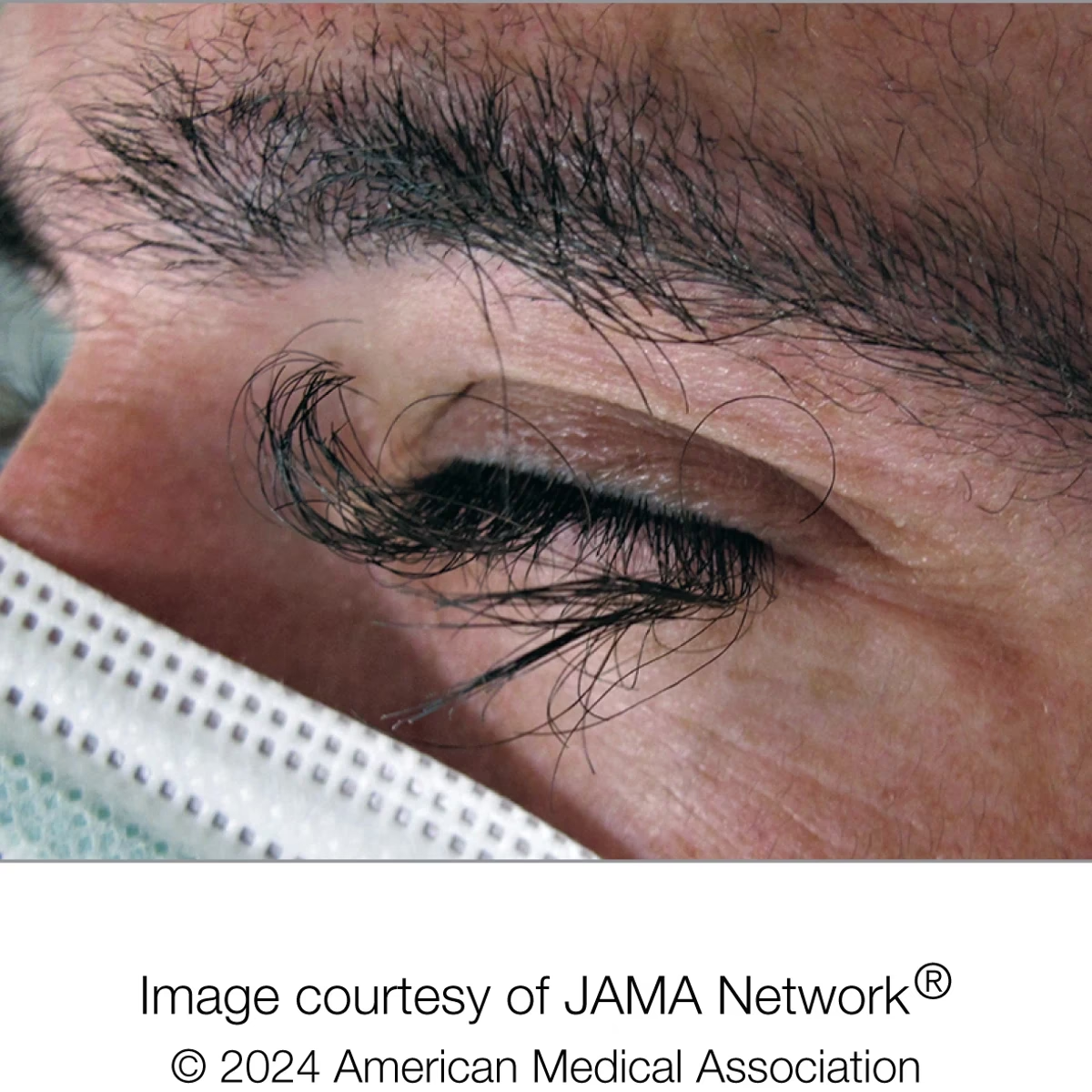
¿Qué es el Síndrome de Cornelia de Lange (CdLS)?
El síndrome de Cornelia de Lange (CdLS) es una condición genética rara y compleja, presente desde el nacimiento, que afecta múltiples aspectos del desarrollo físico, intelectual y conductual. Fue descrito formalmente por primera vez en 1933 por la pediatra holandesa Cornelia de Lange, de quien toma su nombre. Se estima que ocurre en aproximadamente 1 de cada 10,000 nacimientos vivos, lo que lo clasifica como una enfermedad poco común pero significativa por el amplio espectro de sus manifestaciones.
Características Físicas y de Desarrollo del CdLS
Los niños con CdLS suelen nacer con bajo peso y presentan un tamaño y estatura menores de lo esperado para su edad, así como una circunferencia de la cabeza más pequeña de lo normal, una condición conocida como microcefalia. Una de las características más reconocibles y distintivas de este síndrome son sus rasgos faciales particulares, que a menudo incluyen:
- Cejas pobladas que tienden a unirse en el entrecejo (sinofridia).
- Pestañas largas y rizadas, una característica tan llamativa que a veces se le conoce coloquialmente como el "síndrome de pestañas largas".
- Nariz respingona con orificios nasales en posición anterior y un puente nasal deprimido.
- Labios delgados y curvados hacia abajo.
- Orejas de implantación baja.
Más allá de los rasgos faciales, el CdLS puede manifestarse con una serie de otras anomalías físicas y médicas:
- Problemas gastrointestinales: El reflujo gastroesofágico (ERGE) es muy común, lo que puede dificultar la alimentación, provocar dolor y contribuir a un crecimiento lento. También pueden presentarse otras diferencias intestinales.
- Anomalías en las extremidades superiores: Estas varían desde manos pequeñas hasta la ausencia de dedos o de antebrazos, lo que subraya la diversidad de la presentación de la condición.
- Paladar hendido: Una abertura en el techo de la boca, que puede afectar el habla y la alimentación.
- Hernias diafragmáticas: Una condición donde una parte de los órganos abdominales se desplaza hacia el tórax a través de una abertura en el diafragma.
- Problemas de visión y audición: Son frecuentes y pueden requerir intervención temprana para mitigar su impacto en el desarrollo.
- Hirsutismo: Exceso de vello corporal, especialmente en la espalda.
- Defectos cardíacos y problemas dentales: Comunes y pueden necesitar seguimiento médico continuo.
- Convulsiones: Algunas personas con CdLS pueden experimentar episodios convulsivos.
Desarrollo Cognitivo y Conductual en el CdLS
La mayoría de los individuos con CdLS experimentan retrasos en el desarrollo, que pueden variar desde dificultades de aprendizaje leves hasta discapacidades intelectuales profundas. En el ámbito conductual, pueden manifestar comportamientos autolesivos, como morderse los dedos o los labios, y a menudo muestran una preferencia por entornos estructurados, con rigidez e inflexibilidad al cambio, rasgos que pueden asemejarse al espectro autista. También pueden presentarse problemas como el trastorno por déficit de atención con hiperactividad (TDAH).
Causas Genéticas del CdLS
El CdLS es una condición de origen genético. Se ha identificado que diversas mutaciones en genes específicos son la causa subyacente de este síndrome. El primer gen identificado en 2004 por un equipo en el Children’s Hospital of Philadelphia, liderado por Ian Krantz, MD, fue el gen NIPBL. Desde entonces, se han descubierto al menos dos genes adicionales relacionados con el CdLS. En la mayoría de los casos, estas mutaciones no son heredadas de los padres, sino que ocurren de forma espontánea como un cambio de novo en un óvulo o espermatozoide, o muy temprano durante el desarrollo fetal. Sin embargo, existe un pequeño porcentaje de casos en los que no se ha identificado una mutación en los genes conocidos, lo que sugiere la posible existencia de otros genes causantes aún por descubrir.
Pronóstico y Manejo del CdLS
A pesar de la complejidad de sus manifestaciones, una persona con CdLS puede tener una esperanza de vida normal si los problemas médicos asociados, como infecciones recurrentes (especialmente neumonías), problemas intestinales o defectos cardíacos congénitos, son identificados y tratados adecuadamente. El manejo del CdLS requiere un enfoque multidisciplinario que involucre a diversos especialistas para abordar cada una de las necesidades específicas del individuo.

¿Qué es el Síndrome de Noonan?
El síndrome de Noonan es otra afección genética que interrumpe el desarrollo normal en varias partes del cuerpo. Al igual que el CdLS, se caracteriza por un amplio espectro de síntomas que pueden variar de leves a graves, dependiendo del gen específico que contenga la alteración. Esta condición puede afectar los rasgos faciales, la estatura, el corazón y otros sistemas orgánicos. También puede causar que los niños se desarrollen más lentamente de lo habitual en hitos como caminar, hablar o aprender nuevas habilidades.
Síntomas y Características del Síndrome de Noonan
Los síntomas del síndrome de Noonan son muy variados y pueden evolucionar con la edad. Las características faciales, por ejemplo, son más evidentes en la infancia y se suavizan en la adultez. Algunas de las características más comunes incluyen:
Rasgos Faciales
- Ojos muy separados (hipertelorismo), inclinados hacia abajo y con párpados caídos (ptosis). El color de los ojos a menudo es verde o azul claro.
- Orejas de implantación baja y rotadas hacia atrás.
- Nariz con puente hundido, base ancha y punta redonda.
- Boca con un surco profundo entre la nariz y la boca (filtro) y picos amplios en el labio superior. Los dientes pueden estar torcidos, el paladar muy arqueado y la mandíbula inferior pequeña.
- La cara puede parecer tosca en la infancia y volverse más definida con la edad, aunque a veces puede carecer de expresión.
- Cabeza grande con frente amplia y línea capilar baja en la nuca.
- Piel que puede parecer fina y transparente con la edad.
Enfermedad Cardíaca
Muchos individuos con síndrome de Noonan nacen con problemas cardíacos congénitos, siendo la estenosis de la válvula pulmonar el problema más frecuente. Otras afecciones cardíacas incluyen:
- Engrosamiento del músculo cardíaco (miocardiopatía hipertrófica).
- Defectos estructurales como orificios en la pared que separa las cámaras inferiores del corazón (defecto septal ventricular) o estrechamiento de la arteria pulmonar o la aorta (coartación de la aorta).
- Ritmo cardíaco irregular (arritmias).
Problemas de Crecimiento
El síndrome de Noonan afecta el crecimiento normal, lo que lleva a baja estatura en muchos casos. Aunque el peso al nacer suele ser normal, el crecimiento es más lento con el tiempo. Otros problemas incluyen:
- Trastornos de la alimentación que pueden conducir a malnutrición.
- Niveles de hormona de crecimiento que pueden ser bajos.
- Retraso en el estirón de la adolescencia.
- Baja estatura en la adultez, aunque no todas las personas con el síndrome son bajas.
Problemas Musculares y Óseos
- Deformidades en el pecho como pectus excavatum (hundido) o pectus carinatum (proyectado hacia afuera).
- Pezones muy separados.
- Cuello corto, a menudo con pliegues extra de piel (cuello alado).
- Curvatura inusual de la columna vertebral.
Problemas de Aprendizaje, Audición y Visión
La inteligencia de la mayoría de las personas con síndrome de Noonan no se ve afectada, pero existe un mayor riesgo de problemas de aprendizaje y discapacidad intelectual leve. También pueden presentar problemas emocionales y de comportamiento leves. Los problemas oculares son comunes, incluyendo estrabismo (bizquera), problemas de refracción, nistagmo (movimiento rápido de los globos oculares) y cataratas. La audición también puede verse afectada debido a problemas nerviosos o a la estructura ósea atípica del oído interno.
Otros Problemas Comunes
- Problemas de sangrado: Tendencia a moretones y sangrado excesivo debido a una coagulación sanguínea deficiente.
- Trastornos linfáticos: Acumulación de líquido (linfedema), especialmente en manos y pies.
- Afecciones genitales y renales: En hombres, testículos no descendidos es común, lo que puede afectar la fertilidad. La pubertad puede retrasarse en ambos sexos. Los problemas renales suelen ser leves.
- Enfermedades de la piel: Pueden tener problemas en el color y la textura de la piel, así como cabello áspero o escaso.
Causas y Herencia del Síndrome de Noonan
El síndrome de Noonan es causado por un cambio en uno o más genes que producen proteínas que están constantemente activas, interrumpiendo el proceso normal de crecimiento y división celular. Estas alteraciones genéticas pueden ser:
- Hereditarias: Si uno de los padres tiene el síndrome de Noonan y porta el gen alterado, hay un 50% de probabilidad de que cada hijo desarrolle la enfermedad. Esto sigue un patrón de herencia autosómico dominante.
- Casuales (de novo): El síndrome puede desarrollarse en un niño sin antecedentes familiares si la mutación ocurre espontáneamente en un nuevo gen.
En algunos casos, la causa del síndrome de Noonan es desconocida.
Complicaciones y Prevención del Síndrome de Noonan
Las complicaciones pueden incluir retrasos en el desarrollo (que requieren planes educativos individualizados), problemas de sangrado no detectados hasta cirugías, acumulación de líquido (linfedema), infecciones urinarias por anomalías renales, problemas de fertilidad en hombres y un mayor riesgo de ciertos tipos de cáncer, como la leucemia.
La prevención, en términos de evitar la condición, es limitada a menos que haya antecedentes familiares conocidos, en cuyo caso la consejería genética y las pruebas prenatales pueden ser opciones. La detección temprana y la atención médica continua son cruciales para manejar las complicaciones y mejorar la calidad de vida.
Comparación entre el Síndrome de Cornelia de Lange y el Síndrome de Noonan
Aunque ambos son síndromes genéticos que afectan múltiples sistemas corporales y el desarrollo, presentan diferencias clave en sus características, causas genéticas específicas y prevalencia. A continuación, una tabla comparativa:
| Característica | Síndrome de Cornelia de Lange (CdLS) | Síndrome de Noonan |
|---|---|---|
| Prevalencia | Aprox. 1 en 10,000 nacimientos | Más común que CdLS, pero varía significativamente (no se especifica un número exacto en la información proporcionada) |
| Etiología Genética | Mutaciones en genes como NIPBL (el primero identificado), SMC1A, SMC3, RAD21, entre otros. | Mutaciones en genes como PTPN11 (el más común), SOS1, RAF1, RIT1, entre otros. |
| Patrón de Herencia | Mayormente espontáneo (de novo), rara vez heredado. | Autosómico dominante (50% de probabilidad si un padre lo tiene) o espontáneo (de novo). |
| Rasgos Faciales Distintivos | Cejas unidas (sinofridia), pestañas largas, nariz respingona, labios finos, microcefalia. | Ojos muy separados y caídos, orejas de implantación baja y rotadas, cuello corto y alado, pezones separados. |
| Crecimiento | Bajo peso al nacer, estatura y tamaño menores, microcefalia. | Peso normal al nacer, crecimiento más lento, baja estatura común en la adultez. |
| Afectación Cardíaca | Defectos cardíacos congénitos (frecuentes). | Estenosis de la válvula pulmonar (más común), miocardiopatía hipertrófica, otros defectos estructurales. |
| Desarrollo Cognitivo | Retrasos que van de leves a profundos, problemas de aprendizaje. | Riesgo de problemas de aprendizaje y discapacidad intelectual leve, pero la inteligencia no siempre está afectada. |
| Problemas Conductuales | Comportamientos autolesivos, rigidez, preferencia por estructura. | Problemas mentales, emocionales y de comportamiento, generalmente leves. |
| Problemas de Extremidades | Diferencias en extremidades superiores (manos pequeñas, dedos o antebrazos ausentes). | Menos énfasis en anomalías de extremidades, más en deformidades torácicas y esqueléticas. |
| Otros Síntomas Comunes | ERGE, hirsutismo, convulsiones, problemas de visión/audición, paladar hendido. | Problemas de sangrado/coagulación, linfedema, problemas renales, testículos no descendidos, problemas de piel. |
Preguntas Frecuentes sobre Síndromes Genéticos
¿Ambos síndromes son hereditarios?
El síndrome de Cornelia de Lange es predominantemente el resultado de mutaciones genéticas espontáneas (de novo), lo que significa que no se hereda de los padres en la mayoría de los casos. Por otro lado, el síndrome de Noonan puede ser hereditario, siguiendo un patrón autosómico dominante (si un padre lo tiene, hay un 50% de posibilidades de que el hijo lo herede), o puede surgir también por una mutación de novo.
¿Cuál es la esperanza de vida de las personas con CdLS o Síndrome de Noonan?
Las personas con síndrome de Cornelia de Lange pueden tener una esperanza de vida normal, siempre y cuando los problemas médicos asociados, como las infecciones recurrentes, los problemas intestinales o los defectos cardíacos congénitos, sean diagnosticados y tratados de manera adecuada y oportuna. De manera similar, para el síndrome de Noonan, la esperanza de vida puede ser normal si las complicaciones, especialmente las cardíacas, se manejan eficazmente. El monitoreo y la atención médica continua son fundamentales para ambos.
¿Existe una cura para estos síndromes?
Actualmente, no existe una cura para el síndrome de Cornelia de Lange ni para el síndrome de Noonan, ya que son condiciones genéticas. Sin embargo, el tratamiento se centra en el manejo de los síntomas y las complicaciones asociadas. Un enfoque multidisciplinario, que involucra a especialistas en diversas áreas médicas (cardiología, gastroenterología, neurología, terapia física, ocupacional y del habla, genética, etc.), es esencial para mejorar la calidad de vida de los individuos afectados y abordar sus necesidades específicas.
¿Cómo se diagnostican el CdLS y el Síndrome de Noonan?
El diagnóstico de ambos síndromes a menudo se basa inicialmente en la observación de las características físicas distintivas y los retrasos en el desarrollo. La confirmación se realiza mediante pruebas genéticas que identifican las mutaciones en los genes asociados. En algunos casos, si hay antecedentes familiares, se pueden realizar pruebas prenatales. Es importante consultar a un profesional de la salud o a un pediatra si se sospecha de alguna de estas condiciones, quienes pueden remitir al paciente a un genetista u otros especialistas.
¿Qué tipo de especialistas tratan estos síndromes?
Debido a la naturaleza multisistémica de ambos síndromes, se requiere un equipo médico diverso. Este puede incluir pediatras, genetistas, cardiólogos, gastroenterólogos, neurólogos, oftalmólogos, otorrinolaringólogos, endocrinólogos, terapeutas físicos, ocupacionales y del habla, y psicólogos o psiquiatras. La coordinación entre estos profesionales es clave para proporcionar una atención integral y personalizada.
Conclusión
Los síndromes de Cornelia de Lange y Noonan son ejemplos complejos de cómo las alteraciones genéticas pueden influir profundamente en el desarrollo humano. Aunque presentan desafíos significativos, el conocimiento sobre sus características, causas y el manejo adecuado con un enfoque multidisciplinario ha permitido mejorar sustancialmente la calidad de vida y el pronóstico de las personas afectadas. La investigación continua en genética y medicina personalizada ofrece una promesa constante para futuras mejoras en el diagnóstico y tratamiento de estas y otras condiciones genéticas.
Si quieres conocer otros artículos parecidos a Síndromes Genéticos: Cornelia de Lange y Noonan puedes visitar la categoría Cabello.